Chang Liu1,
Fang Liang2,
Yan Li3,
Qiu-Li Wang4 ![]()
For correspondence:- Qiu-Li Wang Email: wangqiuli24@gmail.com Tel:+8637167690392
Received: 13 May 2015 Accepted: 22 December 2015 Published: 28 February 2016
Citation: Liu C, Liang F, Li Y, Wang Q. Mutational analysis of GATA4 and NKX2.5 genes in dilated cardiomyopathy patients. Trop J Pharm Res 2016; 15(2):285-292 doi: 10.4314/tjpr.v15i2.9
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose:To evaluate the lipid profile abnormalities and association of the GATA4 (1232 C→T) and NKX 2.5 (73C→T) gene polymorphisms with Dilated cardiomyopathy (DCM) among Chinese population.
Methods:The blood samples were collected from the Outpatient Department (OPD) of Cardiology Unit, Zhengzhou Central Hospital, China for a period of three years from 2010 – 2013. Dilated cardiomyopathy cases (n = 270) and healthy controls (n = 290) were genotyped using polymerase chain reaction (PCR), restriction fragment length polymorphism, (RFLP) denaturing high-performance liquid chromatography (DHPLC) and sequencing.
Results:Evaluation of the age and sex of the patients indicated that DCM was more prevalent among males (71.1 %) than in females (28.9 %). Mean (± SD) values of the serum total cholesterol and serum LDL-cholesterol were higher in DCM patients (4.37 ± 0.16 and 3.19 ± 0.14 mmol/L, respectively) compared to the control group (4.29 ± 0.18 and 3.06 ± 0.19 mmol/L, respectively), while as there was significantly lower serum mean (± SEM) HDL-cholesterol levels in patients with DCM than in controls (p < 0.001). Exon 1 region of NKX 2.5 gene was screened for variations at 73C→T keeping in view both parameters (age and sex) among healthy controls and DCM patients. The results indicate that the homozygous mutant (TT) and heterozygous mutant (CT) nucleotides were significantly higher in DCM patients than in controls. Further analysis of GATA4 gene revealed that five DCM patients had 1232C/T variant, whereas, it was absent in the control group.
Conclusion:GATA4 1232C/T and NKX 2.5 73C→T polymorphisms and high levels of serum triglycerides (TG) may be associated with the pathogenesis of DCM in the studied population
Introduction
Cardiomyopathies are the heterogeneous group of myocardial disorder in which heart muscle are inflamed, weakened and enlarged leading to heart failure. They are either confined to the heart or become as a part of generalized systemic disorders, often leading to progressive heart failure related disability or cardiovascular death. Dilated cardiomyopathy (DCM) is the third leading cause of heart failure which is ultimately responsible for morbidity and mortality [1]. In more severe cases, DCM is associated with congestive heart failure (CHF), which arises when the heart is unable to pump sufficient blood to meet the body tissue needs for oxygen and nutrients. In most of the cases genetic factors are responsible for development of DCM, whereas, functional characterization of these genetic variants indicated that the diverse cardiomyocyte structural and signalling defects can result in DCM phenotype. Earlier studies have reported that the cardiac transcription factor GATA4 and NKX 2.5 are essential for cardiac development, and mutations in these genes have been implicated in a wide variety of congenital heart diseases in both animal models and humans [2] The expression of these transcription factors extend a large developmental window of cardiogenesis and continues in mature heart. However, whether this mutated GATA4 and NKX 2.5 predisposes to sporadic DCM remains unclear.
ATA4 (MIM 600576) gene encodes a zinc finger containing cardiac transcription factor, it plays an essential role in promoting development and differentiation of the myocardium, as well as in regulating survival and hypertrophic growth of the adult heart. Regular function of GATA4 is essential for cardiac morphogenesis. In vitro and in vivo studies in rats and mice have confirmed the role of GATA4 in cardiac hypertrophy [3].
During fetal development GATA4 is expressed in yolk sac endoderm and cells that are involved in heart formation. It recognizes a consensus sequence known as ‘GATA’ motif, which is an important cis-element in the promoters of many genes. Human GATA4 is located on Chromosome 8 (8p23.1-p22) and consists of 6 exons [3]. GATA4 is expressed in the heart, gut epithelium and gonads. It is highly expressed in cardiomyocytes at different developmental stages and continues to express in the adult cardiac myocytes, where it regulates the transcription of several structural and regulatory genes, including atrial natriuretic factor (ANF), brain natriuretic factor, carnitine palmitoyltransferase Ib, troponin I, troponin C, a- and b-myosin heavy chain which are of vital importance.
NKX2.5 is another essential transcription factor expressed in cardiac precursor cells, crucial for heart development and determination of myocardiocytel fate. The functional significance of NKX 2.5 gene for heart formation and development was observed both in animals and humans. The NKX 2.5 gene is localized to chromosome 5q34 and consists of 2 exons 510bp and 1075bp in size which encodes a 324 amino acid protein. Mutations in NKX2.5 gene have been implicated in diverse congenital heart diseases (CHD), cardiomyopathies and conduction defects (CD) in mouse models and humans. The transcription factor NKX 2-5 is known to have a role in heart development whereas its role in adult-onset heart disease is unknown. Hence, the present study was designed to assess the role of GATA4 and NKX 2.5 gene mutations in dilated cardiomyopathy patients.
Methods
Selection criteria for patient and control group
Two hundred and seventy patients with dilated cardiomyopathy (n = 270), that were referred from the Department of Cardiology, Zhengzhou Central Hospital, Zhengzhou University, Zhengzhou China from 2010 – 2013, were recruited randomly for the study group. Two hundred and ninety (n=290) healthy volunteers corresponding to DCM patients in age and sex but with no family history of DCM and other associated genetic diseases were taken as control group. The study was carried out in accordance with the ethical standards approved by ethics committee of the Department of Cardiology, Zhengzhou Central Hospital, affiliated to Zhengzhou University, Zhengzhou, China. Informed and educated consent to participate in the study was taken from the DCM patients and healthy controls. Clinical diagnosis of DCM was based on the two dimensional echocardiography (ECG), demographic data, clinical features and other associated abnormalities. DCM patients within the age group of 40-50 years were included in the study, while as healthy volunteers corresponding to the same age group with no medical history of either heart diseases or other associated disorders were taken as control.
Collection of blood samples
Blood sample (5 mL) was collected from each subject for biochemical analysis and molecular screening of GATA4 and NKX2.5 gene mutations. Blood samples from the healthy controls were also collected for the analysis and comparison. All the samples were collected after prior description and consent.
Measurement of biochemical variables
Fasting blood samples were collected in liquid EDTA blood tubes, and carried on ice to laboratory, centrifuged, and aliquoted for storage in −80 °C. Fasting total cholesterol, triglyceride (TG), low and high density lipoprotein - cholesterol (LDL-C, HDL-C) were measured in serum of all DCM patients and healthy controls. The total serum cholesterol was determined by the enzymatic colorimetric method of Allain et al [4]. Demacker et al enzymatic method was used for the estimation of serum HDL-C while as serum LDL-C was estimated by using Anwar et al formula [5]. Serum TG was determined according to the enzymatic method of Bucolo and David [6].
Genomic DNA isolation
Genomic DNA was isolated by phenol-chloroform method described by Blin and Stafford [7]. In brief, WBCs were isolated from blood samples using erythrocyte lysis buffer (20 mg/mL Proteinase K, 155 mM ammonium chloride, 7.23 mM potassium carbonate, 0.5 M EDTA, and 20 % SDS) and incubated at 37 oC. Then 5 mL of Tris equilibrated phenol (pH = 8) was added to the sample and centrifuged at 1000 g for 10 min. To the supernatant phenol: chloroform: isoamyl alcohol (25:24:1) mixture was added (5 mL) and incubated for 2 min and then centrifuged at 1000 g for 10 min. Furthermore chloroform: isoamyl alcohol (24:1) was added to the supernatant (5 mL) and centrifuged for 15 min at 1000 g. DNA was precipitated with absolute alcohol (chilled) and dissolved in Tris-EDTA buffer and stored at -20 oC for further analysis.
Testing for sequence variants
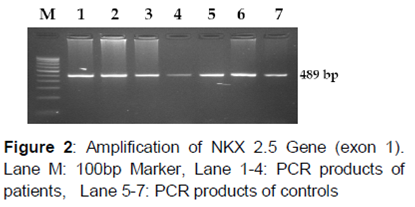
NKX2.5Exon1 (489bp) of NKX2.5 gene was amplified by using forward primer -5’GTCCCGCCTCTCCTGCCCCTTGTG-3’andreverseprimer -5’AGGGTCCTTGGCTGGGTCGG3’, in 50 μL containing 20 pmol each along with 10 mM dNTPs, 3U Taq DNA polymerase, 100 ng of genomic DNA and 10X PCR buffer. The gene was amplified in MJ mini thermal cycler with the subsequent PCR conditions: the initial denaturation was at 95 °C for 7 min, followed by 34 cycles of denaturation (95 °C for 30 sec), annealing (64.5 °C for 1min), extension (72 °C for 1 min) and the final extension step was set for 72 oC for 10 min.
Restriction fragment length polymorphism
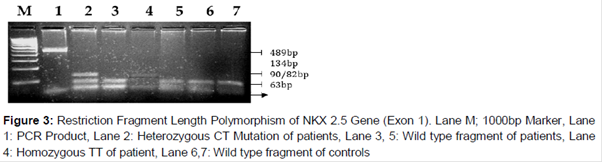
The PCR products from the patients and controls were analyzed for the variant 73C→T in the NKX2.5 gene using HhaI restriction enzyme for RFLP analysis. The reaction mixture of restriction enzyme digest includes restriction buffer, HhaI (3 U), PCR product (10 µl) and sterile water (20 µl). The reaction mixtures were incubated at 65 oC for 3 h. The restriction digestion contents were then resolved on a 12 % PAGE (Acrylamide: bisacrylamide solution-29:1, 10 % glycerol, 10 % APS, TEMED, 10XTBE buffer) followed by staining (20 μg/mL) with ethidium bromide. The resulting restriction digestion yields 134, 90/82 and 63 in heterozygous (CT), 134 and 63 in homozygous (TT) genotypes, 90/82 and 63 DNA fragments in wild type (CC) genotypes, respectively.
GATA4
For GATA4 gene, the coding region was segmented into exon–intron junction sequences suitable for DHPLC (denaturing high-performance liquid chromatography - WAVE, Transgenomics Inc, USA) or CSGE analysis. The 6th exon was amplified from genomic DNA using Forward - 5’ CCTAGACCTCCCAAGC 3’ Reverse 5’GGCCTCCTTCTTTGCTATCC 3’ primers. The PCR containing 20 ng genomic DNA was amplified in a 30 µl reaction mixture containing 1.5 U HotStar Taq polymerase and 16 HotStar Taq PCR Buffer (Qiagen Inc., California, USA), 125 mmol/l of each dNTP (Roche, Germany), 1.5 mmol/l MgCl2, and 1 mmol/l of each PCR primer. The reactions started with 5 min at 95 oC followed by 37 – 40 cycles of 45 s at 95 oC, 30 sec at 61 oC and 45 sec at 72 oC, and finished with a 15-min extension period at 72 oC. PCR products were then examined for sequence alterations by DHPLC and sequencing analysis. Samples that showed peak or band shifts were re-amplified from the genomic DNA and sequenced in both directions with an automated cycle sequencer (ABI BigDye Taq FS Terminator V.3.1; ABI Prism 3100sequencer, Applied Biosystems, Foster City, California, USA).
Statistics
Statistical analysis was performed with Plink software. For evaluation of markers, whether they are in hardy Weinberg equilibrium, and for association analysis, a cut-off p < 0.01 was used. The significance was assessed by Analysis of variance (ANOVA) and Student t-test. The χ2 test was applied to determine the cited equilibrium. Logistic regression analysis was employed to determine the risk of dilated cardiomyopathy, including age and sex as intervening variables. P < 0.05 was considered significant. The final results were shown as odd ratio with the respective 95 % confidence interval (CI).
Results
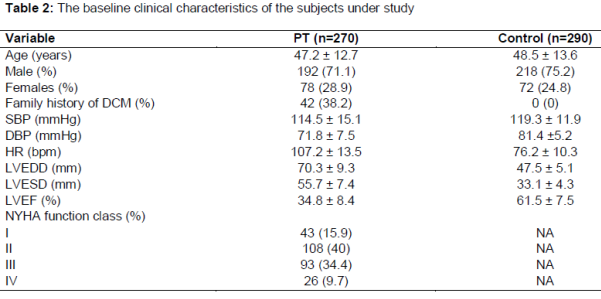
Two hundred and seventy (n=270) dilated cardiomyopathy (DCM) patients and two hundred and ninety (n = 290) healthy controls were analyzed for the variation in lipid profile and gene polymorphisms in NKX2.5 73C→T by RFLP-PAGE using Hha1 and GATA4 by DHPLC/sequencing methods (). Characteristics of the DCM patients and controls are presented in .
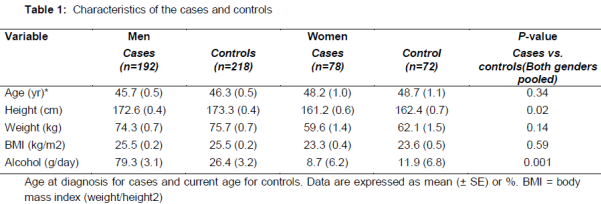
The mean age at diagnosis was 45.7 years in male cases and 48.2 years in female cases. Compared to the controls, the cases were shorter by approximately 1 cm (p < 0.05) and had higher alcohol consumption habits (p < 0.001) (). Transplanted patients were younger in age, leaner and had lower alcohol consumption habits than the non-transplanted patients, and they were characterized by a more severe left ventricular dysfunction and a lower ejection fraction (data not shown).
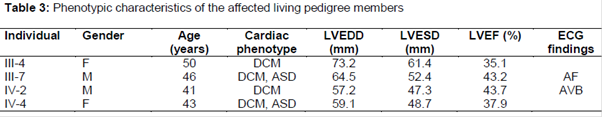
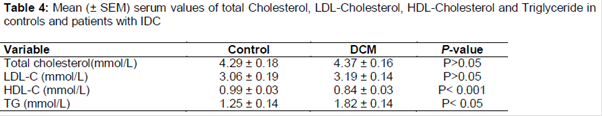
The evaluation of the age and sex frequency of the patients with DCM indicate that DCM was more prevalent among males (71.1 %) than in females (28.9 %). The general characteristics of DCM patient’s associated heart defects and inheritance are represented in the -3. The mean (± SD) values of the serum total cholesterol and serum LDL-cholesterol were found to be higher in the DCM patients (4.37 ± 0.16 mmol/L; 3.19 ± 0.14 mmol/L, respectively) than in the control group (4.29 ± 0.18 mmol/L; 3.06 ± 0.19 mmol/L, respectively) (Tables 1 and 2).
There was a significantly low serum mean (± SEM) HDL-cholesterol level in patients with DCM against the controls (0.84 ± 0.03 mmol/L, 0.99 ± 0.03 mmol/L; p < 0.001, respectively, ).
The mean (± SEM) serum TG levels in DCM patients (1.82 ± 0.14 mmol/L) were significantly higher compared to normal controls (1.25 ± 0.14, p < 0.05) ().
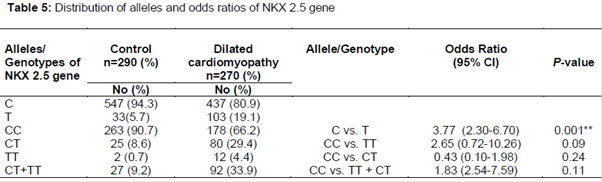
In the present study, exon 1 region of NKX 2.5 gene was screened for variation 73C→T in both age-sex matched healthy controls and DCM patients. The results indicated that the nucleotide variation of NKX 2.5 gene is significantly associated with the DCM patients compared to the controls ().
In , the PAGE gel picture represents the presence of all three genotypes in which the mutant genotypes are more specific for DCM. The NKX2.5 73C→T mutation results in the substitution of Arg25Cys (arginine to cystein) at residue 25 of the protein.
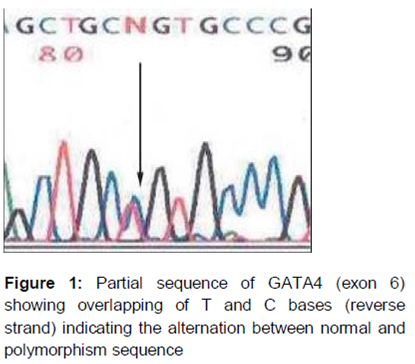
Furthermore, we also investigated the genomic DNA of the DCM patients for variations in exon 6 of the GATA4 gene. Our analysis revealed that the five DCM patients showed 1232C/T variant, whereas it was absent in the controls resulting in Ala411Val change in the protein. The patients carrying the 1232C/T variant did not show family history of cardiomyopathy or any other heart defects.
In addition we investigated whether the polymorphisms were associated with the severity of the disease when assessed by the ejection fraction (,/$24 %), left ventricular dilation (end-diastolic diameter ,/$40 mm/m2) or heart transplantation. The genotype and allele frequencies did not differ between patients with or without a familial history of DCM. As we know that alcohol intake is a major etiologic factor for DCM, we tried to investigate whether alcohol may modulate the effect of polymorphisms on the risk of DCM, but we did not find any evidence to prove, alcohol–genotype interaction.
Discussion
Dilated cardiomyopathy (DCM) is a myocardial disorder that leads to congestive heart failure and sudden cardiac death. It is an indication for cardiac transplantation in patients due to ventricular chamber enlargement and contractile dysfunction [8]. Nearly 50 % of DCM cases are idiopathic and 20-30 % cases are familial. Functional characterization studies indicated that the genetic and environmental risk factors play an important role in the pathogenesis of DCM.
Lipids play an important role in cardiovascular disease complications and there are very few studies on lipid abnormalities in the prevalence of DCM. The observations of the present study indicated that the dilated cardiomyopathy patients had significantly lower HDL-C level and markedly higher TG concentrations than healthy subjects and the results are consistent with the reports of Cooke et al who demonstrated that the mean serum HDL cholesterol serum levels of congestive heart failure (CHF) patients were significantly lower than that of normal subjects (0.86 ± 0.24 and 1.2 ± 0.31 mmol/L, respectively). The present study is in contrast with the results of Freitas et al [9] who reported a higher serum HDL cholesterol levels in heart failure patients.
Similarly, Basil et al [10] also observed no statistical differences between the serum total cholesterol and LDL-cholesterol levels of the idiopathic dilated cardiomyopathy patients compared to the normal controls. Elevated serum LDL-cholesterol levels are fundamentally related to ischemic heart disease (IHD) and its reduction reduces the incidence of IHD and the associated mortality rate. Basil et al [10] also reported that HDL-cholesterol functions as an autonomous protective factor for the endothelium. It has been observed that HDL-cholesterol induces the activation of endothelial NO synthase, NO release, and vasorelaxatory effects [8,11]. Earlier studies have reported that increased levels of serum triglycerides are associated with heart failure and our results also demonstrates a statistically significant relationship between serum triglycerides and mortality [12,13].
Several genetically engineered models for dilated cardiomyopathy have been reported recently, to explain the role of genes/transcription factors in the development of the disease [14]. A recent study has implicated GATA4 gene mutation in the pathogenesis of familial DCM [15,16]. Functional analysis demonstrates that the GATA4 mutation was significantly associated with the decreased transcriptional activity and remarkably reduced synergistic activation between GATA4 and NKX2-5, another transcription factor is crucial for cardiogenesis and the functioning of the heart [17]. GATA4 may bind to the target DNA in association with other partners including NKX2-5, and the synergistic transcriptional activation mediated by NKX2-5 has been verified [18,19]. However there are no reports on the GATA4 1232 C→T and NKX2.5 73C→T gene polymorphisms yet. Therefore, the present study was carried out to analyze the frequency of GATA4 and NKX 2.5 gene polymorphisms in patients suffering from dilated cardiomyopathy. The current study indicates that the five DCM patients had 1232C/T variant, whereas it was absent in the control group (). The patients carrying the 1232C/T variant did not show family history of cardiomyopathy or any other heart defects. Among the various polymorphisms of the GATA4 gene, it is now evident that in exon 6, the 1232C/T variant has important implications in the etio-pathogenesis of cardiovascular diseases and DCM being a complex disorder associated with heart failure may be one of such CVD with the underlying mechanism of oxidative stress.
GATA4 is a transcription factor that belongs to the GATA family of zinc finger proteins expressed in various organs including the hematopoietic system, the heart, the gonads, the lungs, and the kidneys [20]. The C-terminal finger acts as a DNA-binding domain and it interacts with other transcription factors such as NKX2.5 and TBX5. The N-terminal domain helps in stabilizing DNA binding [19,21]. In vitro and in vivo studies in rats and mice have confirmed the role of GATA4 in cardiac hypertrophy. Loss of function mutation of GATA4 in mice resulted in their inability to undergo physiological hypertrophy during pressure overload.
GATA4 is also found to be associated with hypertrophic cardiomyopathy caused by cardiomyocyte hyper-proliferation without hypertrophy [14] In a recent study, Li et al [16] reported an autosomal dominant missense mutation (c.812G > C) that results in a p.C271S substitution in the amino acid sequence in DCM patients with 100 % penetrance. Similarly, Zhao et al [1] also demonstrated an autosomal dominant novel heterozygous GATA4 mutation (p.V291L) in DCM.
Cardiac transcription factors GATA4 and NKX 2.5 are the essential transcriptional factors expressed predominantly in the heart and they regulate the expression of the cardiac genes encoding structural proteins [22]. Zhao et al [1] reported genetic polymorphisms in GATA4 and NKX2.5 genes as potential transcriptional factors of hypertrophic and dilated cardiomyopathy phenotypes. Yuan et al [23] observed a novel NKX2-5 loss-of-function in familial dilated cardiomyopathy and arrhythmias patients. Similar findings were observed in the present study. An increased frequency of 73C→T variant was detected in the DCM patients. The prevalence of variation in the present study was found to be 4.4 % in homozygotes (TT) and 29.4 % in heterozygous patients (CT) compared to the controls (TT-0.7 %: CT-8.6 %). Similarly, Yuan et al [23] also reported p.S146W polymorphism in a family with DCM which was inherited as an autosomal dominant trait and co-segregated with DCM in the family with complete penetrance. Pashmforoush et al [24] observed that the Nkx2-5 mutations leads to progressive cardiomyopathy and conduction defects.
NKX2-5 is crucial for heart development and mutations in this gene is implicated in diverse congenital heart diseases and conduction defects in mouse models and humans [7]. NKX2.5 is the earliest known marker for myocardial progenitor cells in all species in which it has been studied. Previous reports have described the NKX2.5 mutations in various congenital heart defects [25]. The nucleotide variation 73C→T corresponds to change from arginine to cysteine (Arg25Cys) at residue 25 in the protein of NKX2.5 transcription factor resulting in the charge of an amino acid from basic to neutral and is highly conserved between species. Arg25Cys is a missense mutation and is present outside the homeodomain region in the amino terminus of the non-homeodomain region. Arg25 residue does not lie within a functional domain of NKX2-5, according to secondary structure prediction with the protein forecasting (PROF) algorithm. Arg25 is just outside the first helix region of NKX2-5 spanning residues 14 to 24. The highly charged nature of arginine also dictates solvent accessibility. Mechanistically, NKX2.5 seems to be regulating precise target gene activation at each developmental stage and the mutations in NKX2.5 could not bind to the other transcription factors leading to cardiac abnormalities including DCM.
Conclusion
The present study indicates that GATA4 and NKX2.5 mutations may be penetrant with variable expressivity in the DCM patients. The higher frequency of the mutant alleles/genotypes and the abnormal lipid profile in the present study may be due to genetic diversity and ethnic variation. Hence, GATA4 and NKX2.5 are possible candidate genes in the athogenesis of DCM. Furthermore, identification and screening of mutations will provide insight into the potential role of the GATA4 and NKX2.5 genes in the development of dilated cardiomyopathy in patients.
Declarations
Acknowledgement
References
Archives


News Updates